Фазовый анализ полезных ископаемых
Фазовым анализом называется совокупность физических и химических методов, позволяющих изучить распределение любого элемента по компонентам изучаемого объекта. Первая задача фазового анализа — определение формы нахождения элемента, то есть диагностика минеральной формы (фазы), с которой связан элемент, и определение ее содержания. Вторая задача — определение фазового состава, то есть диагностика всех минеральных фаз с количественной оценкой их содержания.
Фазовый состав горной породы и особенно количественное соотношение минеральных фаз определяют технические характеристики горной породы, ее технологические свойства. Это особенно важно, если порода является рудой. Полная фазовая минеральная характеристика обеспечивает комплексное использование минерального сырья.
Следует различать частичный (частный) и полный анализ фазового состава минеральных объектов. При частичном фазовом анализе определяются минеральные формы нахождения одного элемента и количество одной или нескольких его минеральных форм в пробе. При полном анализе проводится диагностика и оценка содержания всех минеральных фаз руды или продуктов ее технологической переработки.
Существуют методы полного количественного фазового анализа и методы ограниченных возможностей. К первым относятся методы оптической микроскопии и рентгенографии, ко вторым — термического анализа (диагностирующего только термоактивные фазы), магнитометрия (анализирующая фазы, содержащие в своем составе железо, марганец и некоторые другие парамагнитные ионы), ИК-спектроскопия, люминесценция (анализирующая только люминесцирующие фазы), мессбауэровская спектроскопия (определяющая фазы Fe и Sn), квантово-оптическая спектроскопия (выявляющая фазы без центра симметрии), химический фазовый анализ и другие.
Метод оптической микроскопии позволяет проводить фазовый анализ с помощью оптического микроскопа. Анализ основан на определении площади шлифа, занимаемой каждой из присутствующих в нем предварительно диагностированных минеральных фаз (оптико-геометрический анализ). Для получения надежных результатов необходим усредненный набор (20—25) статистических данных по большому количеству шлифов одного минерального объекта. Это делает анализ весьма трудоемким и порождает стремление к его автоматизации. Условием автоматизации является стабильность оптических свойств (цвет, поглощение, отражение) каждой определяемой фазы хотя бы в пределах анализируемой пробы. Путем настройки детекторов на определенные оптические характеристики анализируемых фаз и последующего сканирования световым лучом площади объекта осуществляется оценка содержания каждой минеральной фазы в пробе.
Технические средства этого метода позволяют проводить фазовый анализ как в полуавтоматическом (визуальная регистрация), так и в полностью автоматическом (фотоэлектрическая регистрация) режимах. Время полного анализа одного шлифа при одновременной визуальной регистрации 10 минеральных фаз, например, на приборе МИУ-1 (микроскопическое интегральное устройство), в среднем составляет 20—25 мин при воспроизводимости анализа ±1 %.
При использовании автоматических микроскопических устройств («Квантимет», «Контраст» и т.д.) полный анализ возможен лишь в благоприятных случаях, то есть когда анализируемые фазы обладают достаточной контрастностью относительно друг друга. Чаще автоматический режим применяют для выполнения неполных анализов. Время одного анализа составляет от нескольких секунд до 3 мин (прибор «Контраст»), воспроизводимость анализа ±0,5 %.
Зарубежные фирмы выпускают для данного вида анализа приборы, в которых фоторегистраторы связаны с ЭВМ. Это обеспечивает относительно высокую экспрессность и точность анализа. Так, анализатор «Квантимет-720» (Англия) проводит одновременное определение содержания в шлифе до 25 фаз, различающихся оптически.
Однако микроскопический анализ успешно осуществляется лишь в том случае, если размеры анализируемых фаз не выходят за пределы разрешающей возможности микроскопа, то есть составляют > 1-0,5 мкм, при условии достаточной контрастности фаз относительно друг друга.
В основе рентгенофазового анализа лежат следующие принципы: порошковая дифракционная картина является специфической характеристикой кристаллического вещества; каждая кристаллическая фаза дает всегда одинаковый дифракционный спектр; рентгенофракционный спектр смеси индивидуальных фаз является суперпозицией (результирующий эффект) их дифракционных спектров; по дифракционному спектру смеси возможна количественная оценка соотношений кристаллических фаз, присутствующих в изучаемом образце.

В результате проведения исследований на дифрактометре получают дифрактограмму (рентгенодифракционный спектр) в виде заданной по точкам спектральной функции, выведенной на цифропечать, перфоленту или в виде графического изображения этой функции на ленте самописца. Современные дифрактометры позволяют получить рентгенодифракционный спектр в диапазоне углов 28 (0 — угол между падающим рентгеновским лучом и отражающей плоскостью) от 6—8 до 140—160° с шагом Д(20) 0,01—0,005°. Если считать, что половина диапазона получения спектра относится к областям фона, то информативные области спектра будут содержать порядка 7,5*10в3—15*10в3 точек. Для решения конкретных аналитических задач 0 проводится предварительная обработка спектра (отделение фона, определение положений пиков (по их максимумам), вычисление интегральных интенсивностей), результатом которой является сжатие спектральной информации примерно на два порядка. Такой сжатый рентгенодифракционный спектр представляется в виде совокупности пар значений {20i, Ii} или {di, Ii}, где 20i или di определяет положение i-той линии в шкале углов 26 или межплоскостных расстояний d, a Ii есть интегральная интенсивность i-ой линии. Графически сжатый спектр представляется в виде штрих-диаграммы (рис. 2.32).
Величины межплоскостных расстояний d определяются из уравнения Вульфа—Брэгга:

где Л — длина волны используемого характеристического излучения, 0 — угол скольжения.
Рентгенофазовый анализ по дифракционным спектрам поликристаллических образцов представляет собой эмпирический метод, включающий количественное сопоставление значений межплоскостных расстояний (d) изучаемого образца с соответствующими значениями d одного или более эмпирических справочных стандартов при качественном сравнении интенсивности линий (I) в спектрах образца и стандартов. Для проведения рентгенофазового анализа неизвестного вещества требуется набор надежных справочных стандартов кристаллических фаз, представленных совокупностью величин d и I. В мировой практике для рентгенофазового анализа широко используется картотека Объединенного комитета дифракционных стандартов (JCPDS).
В зависимости от поставленной задачи и характера объекта на практике применяются различные варианты рентгеновского количественного фазового анализа. Если задачей анализа является определение одного минерала (фазы) или определение соотношения двух минеральных фаз и его изменение в каком-либо процессе, то чаще всего используют метод стандартных смесей и метод добавок, метод внутреннего и внешнего стандарта.
Однако при анализе многокомпонентной системы, содержащей 6—7 и более минеральных фаз, введение дополнительной фазы-эталона является нежелательным, поскольку в довольно сложной дифракционной картине трудно выбрать независимый неусложненный наложением аналитический рефлекс каждой фазы. Эталон дает дополнительные рефлексы. Оптимальным в этом случае является дифракционно-абсорбционный метод, пригодный для анализа любых ассоциаций минералов на любом рентгеновском излучении с минимальным объемом предварительных исследований. Рабочей формулой метода является формула

где Ci — содержание i-го компонента в пробе; uпр* и u0i* — коэффициенты массового поглощения пробы и i-то компонента в чистом виде; Ii и I0-интенсивности опорного (аналитического) рефлекса i-го компонента в пробе и в чистом виде.
Предметом аналитических определений являются замеры u' и I. Предварительный этап работы включает качественный фазовый анализ, получение чистых фаз пробы и оценки их u'.
Методики рентгеновского дифрактометрического анализа позволяют оценить содержание всех раскристаллизованных фаз, величина кристаллов которых > 0,01 мкм (более высокодисперсные образования рентгеноаморфны) и содержание > 1 %. Для обнаружения, диагностики и оценки содержания фаз более низкой концентрации необходимо фракционирование пробы, например, по плотности или другим признакам. Содержание рентгеноаморфных и аморфных фаз оценивается путем вычитания суммы кристаллических фаз из 100 %. Диагностика этих фаз проводится методом ИК-спектроскопии.
Точность определения содержания фазы рентгенодифракционным методом ±(1—2) % абс. и зависит от общего состава пробы и степени раскристаллизации минеральной фазы. Навески для анализа — от 20 до 100 мг в зависимости от задачи исследования.
В настоящее время анализ осуществляется на отечественных дифрактометрах УРС-50ИМ, ДРОН-1 и ДРОН-2, а также АДП-2,0 и ДАРП-УМ с автоматизацией отдельных операций анализа.
Термомагнитометрический метод основан на использовании естественного магнетизма некоторых минералов и является средством диагностики и количественной оценки содержания в пробе сильномагнитных (ферро- и ферримагнитных) минералов. Диагностика магнитной фазы основана на анализе превращений, претерпеваемых минералом при нагревании, то есть структурных преобразований, связанных с разрушением минерала и перекристаллизацией вещества (сидерит—маггемит, маггемит—гематит), и магнитных превращений, обусловленных изменением магнитного состояния при температуре Кюри (магнетит, пирротин и др.), температуре Морина (гематит). Количественный анализ основан на определении намагниченности пробы, пропорциональной содержанию в ней магнитных минералов, так как влияние парамагнитных минералов на суммарную намагниченность пробы незначительно. К числу фаз, анализируемых термомагнитным методом, относятся прежде всего минералы железа: а — Fe, простые и сложные оксиды (вюстит, магнетит и члены его изоморфных рядов — магномагнетиты, титаномагнетиты, франклиниты, якобсит, треворит, хлорит), минералы серии гематит — ильменит, гидроокислы — гетит, гидрогематит, лепидокрокит и др., сульфиды (пирротин, кубанит), карбонаты (сидерит). Объектами исследования могут быть как хорошо раскристаллизованные минералы, так и высокодисперсные, вплоть до рентгеноаморфных. Предел обнаружения — 0,1—1 %. Воспроизводимость ±2 %. Производительность анализа зависит от задач и сложности проб и варьирует от 2 до 20 проб в день. Анализ проводят с помощью вибрационного магнитометра. Увеличение чувствительности, экспрессности и круга анализируемых минералов даст возможность широко применять метод при изучении вещественного состава руд и горных пород.
Ядерный гамма-резонансный спектроскопический метод применяется для диагностики и количественной оценки минеральных фаз, содержащих железо и олово. Метод ЯГРС широко применяют для определения содержания касситерита по резонирующему изотопу 119Sn с использованием портативной аппаратуры типа МАК. Известно около 100 резонансных изотопов более 40 элементов от 40K до 243Am (Sb, Fe, Dy, Au) и более 40 радиоактивных резонансных изотопов, однако регистрация их ЯГР-спектров более сложна и требует разработки индивидуальных методов анализа.
В квантово-оптическом методе для оценки содержания в пробе минералов, обладающих нелинейными оптическими свойствами, то есть минералов, кристаллическая структура которых не имеет центра симметрии, используют лазерное излучение. В основе анализа лежит способность таких минералов генерировать II гармонику лазерного излучения, то есть излучения с удвоенной частотой. Интенсивность генерированной II гармоники определяется прежде всего содержанием нецентросимметричного минерала в пробе, что и используется при количественном фазовом анализе.
К минералам с нелинейными оптическими свойствами относят кварц, нефелин, турмалин, сфалерит, вюртцит, киноварь, миллерит и другие более редкие минеральные виды — всего ~ 260. При содержании в пробе нескольких нецентросимметричных минералов интенсивность второй гармоники дает информацию о суммарном их содержании. Однако в породе чаще преобладает один нецентросимметричный минерал, например, кварц или нефелин. В этом случае метод надежно используют для оценки содержания этого минерала при массовом определении объекта. В настоящее время метод разработан для определения содержания кварца в различных породах. Время выполнения анализа одной пробы — 30 с (исключая пробоподготовку). Это позволяет рекомендовать его для массовых анализов. Относительная воспроизводимость +5—6 %. Предел обнаружения 0,1 %. Установка для анализа монтируется на базе стандартной серийной аппаратуры.
Метод основан на эффекте резонансного взаимодействия у-излучения с ядрами атома кристаллической структуры. Естественно, что с изменением кристаллической структуры изменяется энергия резонансного взаимодействия одного и того же элемента, что позволяет по ЯГР-спектру проводить диагностику минеральной фазы, характеризующейся прежде всего индивидуальной кристаллической структурой. В результате сверхтонких взаимодействий основной энергетический уровень ядра резонансного изотопа железа (57Fe) или олова (119Sn) может сместиться по энергетической шкале, расщепиться на два («квадрупольное расщепление») или шесть (магнитное расщепление) уровней. Используя малую энергетическую ширину резонансной линии, можно, изменяя энергию у-квантов путем перемещения с определенной скоростью источника излучения относительно пробы, создать условия, при которых энергия у-кванта делается равной определенному уровню ядра. Наступает резонансное взаимодействие, в результате которого проба сильно поглощает излучение. Задавая различные скорости перемещения источника относительно пробы и измеряя интенсивность излучения, прошедшего через пробу, можно получить спектр сверхтонких взаимодействий 57Fe, 119Sn. Это ЯГР-спектр пробы, который определяется валентностью иона, характером его химических связей, координацией его в кристаллической структуре, степенью совершенства последней, проявлением изоморфизма и наличием воды. ЯГР-спектры позволяют проводить диагностику минеральной фазы, содержащей резонирующий атом (ион), с оценкой особенностей ее строения.
С помощью этого метода в порошках и рудах по характеру спектра железа выявляют оксиды железа — гематит, магнетит, маггемит, ильменит, гетит, лепидокрокит; карбонаты — анкерит, сидерит, сульфиды и силикаты, в состав которых входит железо. Принимая постоянным содержания железа в любой Fe-содержащей минеральной фазе, по содержанию железа определяют содержание каждой фазы с помощью эталонировочных кривых и стандартных спектров.
Воспроизводимость определения железа в смеси Fe-содержащих фаз составляет в среднем 1 %. Время получения спектра — 40—50 мин. Навеску (от 100 мг до 2 г) измельчают до -0,1 мм.
Экспериментальные работы проводят на серийных приборах ЯГРС-3 или ЯГРС-4.
Химический метод фазового анализа основан на применении селективных растворителей. Разработка методик химического фазового анализа начинается с отбора чистых минералов, их анализа и идентификации различными методами. Далее основные усилия исследователей направляются на подбор селективных растворителей выделенных минералов. В качестве растворителей используют различные растворы кислот, щелочей, солей, комплексообразующих реагентов и т.п. Существенное значение имеет выбор наиболее рациональных условий обработки растворителем: продолжительность обработки, температура, степень измельчения обрабатываемой пробы, отношение массы твердого вещества к массе жидкого (Т:Ж), скорость перемешивания реакционной смеси и др.
В качестве растворителей обычно используются такие реагенты, которые в определенных условиях либо переводят в раствор полностью одну форму соединения элемента и не затрагивают другие, либо растворяют их в сравнительно малой степени (на 5—10 %), либо переводят в раствор одну форму соединения не менее чем на 90 %, а другую — не более чем на 10 %. Такие растворители называются селективными.
Недостаточная избирательность растворителей служит одной из основных причин погрешностей результатов химического фазового анализа. Величина погрешности зависит не только от степени растворения, но и от содержания определяемой формы в анализируемом материале. Наибольшие ошибки в результатах анализа получаются при определении тех фаз, которые содержатся в небольших количествах при относительно больших количествах других.
В химическом фазовом анализе растворение является гетерогенной реакцией, происходящей при взаимодействии двух веществ: реагента-растворителя, находящегося в жидкой фазе, и минерала или соединения, находящегося в твердой фазе (пробе исследуемого материала). В результате такого взаимодействия компоненты минерала образуют с реагентом растворимые в данной среде соединения, которые затем могут быть определены любым методом, применяемым для определения элемента в растворе.
Приступая к фазовому анализу неизученной руды, необходимо на штуфных образцах или мономинеральных фракциях убедиться, что применяемая в других случаях методика анализа пригодна и для данной руды. Для руд сложного состава необходимо определить степень растворения всех соединений изучаемого элемента в отдельности и в смеси друг с другом.
В настоящее время имеется большое количество методик химического фазового анализа, позволяющих определить минеральные формы Pb, Sn, Mo, W, Cu, Mn, Ti, Fe, Cr, Ni, Sb, As, Bi, Nb, Ta, Zn, S и др., а также методы определения растворимого и нерастворимого Be, свободного глинозема и свободной кремнекислоты. Описания этих методов приведены в методических руководствах HCAM и статьях.
Рассмотрим несколько примеров химического фазового анализа некоторых руд.
Пример 1. Определение соединений меди в рудах и продуктах их переработки.
Известно около 179 минералов, содержащих медь. Перечень важнейших медных минералов, имеющих промышленное значение, приведен в табл. 2.29. Халькопирит, кубанит (и иногда борнит) являются первичными сульфидами. Халькопирит встречается почти во всех медных месторождениях руд, часто в тесном срастании с пиритом. Халькозин, ковеллин и борнит являются вторичными сульфидами, образовавшимися при взаимодействии продуктов окисления халькопирита с пиритом и сульфатом меди. Сложные сульфиды меди, содержащие мышьяк As и сурьму Sb (блеклые руды), встречаются реже других сульфидов и главным образом в полиметаллических рудах. Из окисленных минералов меди наибольшее распространение имеют малахит, азурит, куприт и тенорит. В некоторых месторождениях имеется хризоколла.

По минеральному составу и характеру вмещающих пород различают несколько типов медных руд. Так, пластовые руды являются монометаллическими рудами. Медные порфировые руды также монометаллические, однако иногда содержат молибден. Медь в таких рудах представлена халькопиритом, реже халькозином. Основную массу руды (95 %) составляют кремнийсодержащие минералы: кварц и серициты. Колчеданные руды на 95 % состоят из сульфидов железа: пирита, реже пирротина. Медь представлена главным образом халькопиритом. Имеются также медно-никелевые руды, содержащие большое количество пирита, пирротина и пентландита, а также другие руды.
Фазовый анализ смешанных медных руд сравнительно простого минерального состава часто заключается лишь в определении суммарного содержания окисленных минералов меди (то есть меди, содержащейся в руде в виде оксида, карбоната, сульфата и силиката) и сульфидных минералов меди (то есть меди, содержащейся в виде первичных и вторичных сульфидов) (рис. 2.33).
Иногда требуется раздельное определение сульфатной меди, суммарное определение вторичных сульфидов меди (халькозина Cu2S, ковеллина CuS и борнита Cu3FeS4), а также определение первичного сульфида — халькопирита CuFeS2.
Для руд сложного минерального состава приходится вести более подробный анализ (рис. 2.34).
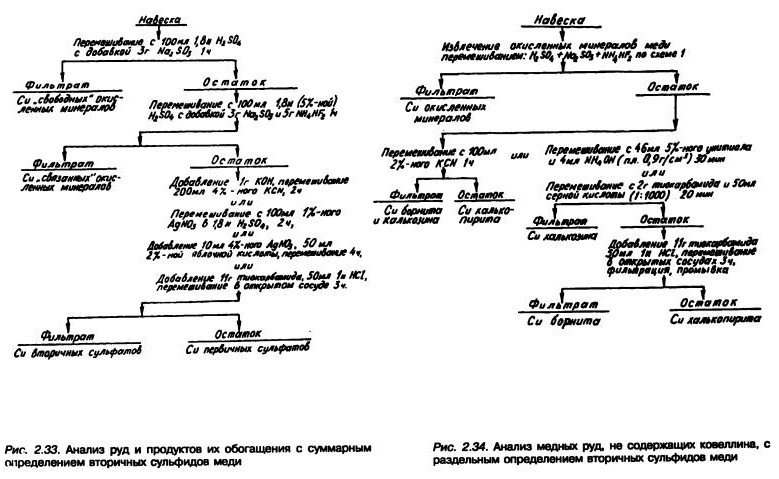
В некоторых рудах содержится хризоколла, которая плохо флотируется и, таким образом, в значительной мере определяет потери меди в хвостах флотации смешанной или окисленной руды. Для определения хризоколлы рекомендовано использовать водный раствор унитиола, который извлекает все окисленные минералы меди, кроме хризоколлы и тенорита, которые затем извлекаются солянокислым раствором унитиола. Силикаты других металлов не переходят при этом в раствор, поэтому, определив в фильтрате медь и кремнекислоту, можно рассчитать содержание обоих минералов.
Более надежным методом определения хризоколлы, хотя и более трудоемким, является выделение этого минерала (плотностью - 2,5 г/см3) в легкую фракцию при обработке руды тетрабромэтаном.
Пример 2. Определение соединений свинца в рудах и продуктах их переработки.
В природе встречается много минералов свинца, представляющих собой различные химические соединения: содержащие серу (сульфид, сульфат), окислы, средние, основные и смешанные соли различных кислот, содержащих W, Mo, V, Р. Перечень важнейших минералов свинца приведен в табл. 2.30.

Наибольшее промышленное значение имеет сульфид свинца — галенит. Непременными спутниками этого минерала являются сфалерит (сульфид цинка) и аргентит (сульфид серебра). Одновременно с галенитом часто встречаются халькопирит и другие сульфиды меди, а также минералы сурьмы, висмута и мышьяка.
Месторождения чистых свинцовых руд встречаются редко. Различают четыре категории свинцовых руд — чисто свинцовые, свинцово-цинковые, свинцово-серебряные и полиметаллические. Последние содержат в промышленных концентрациях несколько полезных компонентов: Pb, Zn, Cu, часто Ni и благородные металлы.
Наибольшее практическое значение имеют полиметаллические руды, в которых свинец представлен главным образом галенитом. В окисленных рудах свинец находится в виде церуссита и англезита, которые часто ассоциируются с карбонатами цинка, меди, железа и других металлов. Вмещающая порода состоит преимущественно из пирита, встречаются также барит, кварц и другие силикаты.
Полная схема фазового анализа смешанной свинцовой руды и хвостов ее флотации приведена на рис. 2.35.
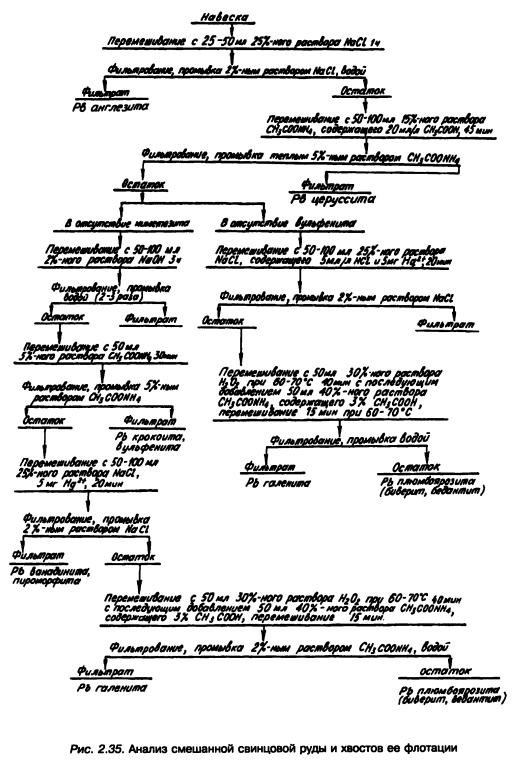
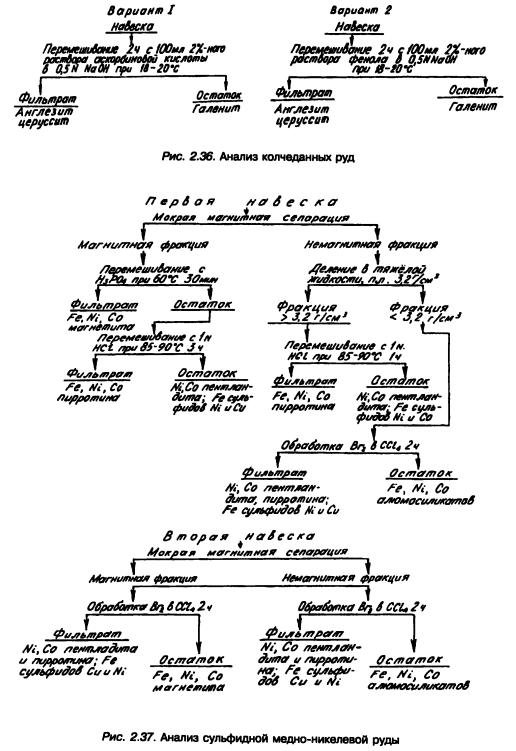
При фазовом анализе колчеданных руд с большим содержанием пирита определение окисленных соединений свинца ведется по двум вариантам схемы (рис. 2.36), точность которых одинакова.
Пример 3. Определение соединений никеля в рудах и продуктах их переработки.
Никель встречается в природе в виде многочисленных и разнообразных соединений. Образуя около пятидесяти самостоятельных минералов, никель входит в состав многих соединений других элементов, изоморфно замещая атомы этих элементов в кристаллической решетке соответствующих минералов или же образуя с ними изоколлоидные смеси высокой дисперсности и соединения адсорбционного типа. Имеются самостоятельные сульфиды никеля; кроме того, сульфид никеля входит в состав пирротина. Наряду с самостоятельными никелевыми окисленными минералами никель входит во многие силикаты (табл. 2.31).

Руды, содержащие никель, делятся на сульфидные и силикатные. Главными компонентами сульфидных руд являются никель и медь, причем никель представлен в таких рудах главным образом в виде самостоятельного сульфидного минерала (пентландита) и в виде изоморфной примеси в пирротине и лишь в небольшом количестве входит в состав силикатов. В таких рудах встречаются теллур, селен, золото, серебро. Одна из схем фазового анализа сульфидной медно-никелевой руды показана на рис. 2.37.
В силикатных рудах никель представлен гидросиликатами никеля и магния, никельсодержащими водными оксидами железа и алюминия. В оливине, серпентине и других силикатах никель содержится в виде оксида, изоморфно замещающего MgO.
Фазовый анализ силикатных никельсодержащих руд основан на различном отношении силикатов к сильным кислотам. Оливин чрезвычайно легко разлагается сильными кислотами; пироксены не разлагаются даже при длительном кипячении; никелевые силикатные минералы по своей кислотостойкости занимают промежуточное положение.
Пример 4. Определение соединений железа в рудах и продуктах их переработки.
Железо в сульфидных рудах цветных металлов находится главным образом в виде простых (пирит, пирротин) и сложных сульфидов с различными цветными металлами: Cu (халькопирит, борнит), Ni (пентландит), As (арсенопирит). В смешанных и окисленных рудах находятся окисленные минералы железа: силикаты, водные оксиды, сидерит, магнетит. Определение форм железа и его общего содержания проводят по схемам, приведенным на рис. 2.38 и 2.39, а на рис. 2.40 приведена схема фазового анализа золота.
Существующие методы химического фазового анализа не могут претендовать на точность обычного химического анализа и позволяют проводить определение минеральных форм с относительной воспроизводимостью ±1—10 %. Однако химический фазовый анализ при всей трудоемкости и ограниченности его методик не может быть исключен из употребления из-за высокой его чувствительности при выявлении фаз низкого содержания, недоступных физическим методам.
Результаты определения распределения и форм нахождения ценных элементов в рудах оформляют в виде таблиц (см. табл. 2.32—2.33).


Полученные данные о вещественном составе руды представляются в виде отчета. Отчет должен содержать задачи исследования, описание материалов для исследования и краткие данные о предыдущих исследованиях вещественного состава руд данного месторождения; краткую геолого-минералогическую характеристику месторождения; методы отбора и обработки пробы; количественный химический состав; качественные и количественные данные о минеральном составе с макро- и микроописанием основных и ценных минералов; распределение ценных элементов по минералам руды по данным химического, спектрального и минералогического анализов; заключение; список использованной литературы.
Следует отметить, что исследования вещественного состава технологических проб полезных ископаемых выполняются геологами, физиками и химиками. Однако обогатители должны знать о том, как получается информация о вещественном составе руды, и уметь оценить достоверность и надежность этой информации.

Другие новости по теме:
Информационный некоммерческий ресурс fccland.ru ©
При цитировании информации ссылка на сайт обязательна.
Копирование материалов сайта ЗАПРЕЩЕНО!
При цитировании информации ссылка на сайт обязательна.
Копирование материалов сайта ЗАПРЕЩЕНО!